Insulin Resistance
Insulin resistance is present when the biological effects of insulin are subnormal for both glucose disposal in skeletal muscle and suppression of endogenous glucose production primarily in liver.
In the fasting state, however, muscle accounts for only a small proportion of glucose disposal (less than 20%) while endogenous glucose production is responsible for all of the glucose appearing in plasma.
In patients with type 2 diabetes and in patients with impaired fasting glucose (IFG) endogenous glucose production is accelerated.
Since many of these subjects may (still) have basal hyperinsulinemia, at least early in the disease, hepatic insulin resistance (with increased hepatic glucose production) is the driving force of hyperglycemia of type 2 diabetes.
 FIGURE 1 Hyperbolic relationship between beta-cell function and insulin sensitivity. In subjects with normal glucose tolerance a quasihyperbolic relationship exists between beta-cell function and insulin sensitivity. With deviation from this hyperbola, deterioration of glucose tolerance. Abbreviations: IGT, impaired glucose tolerance; NGT, normal glucose tolerance; T2DM, type 2 diabetes mellitus.
FIGURE 1 Hyperbolic relationship between beta-cell function and insulin sensitivity. In subjects with normal glucose tolerance a quasihyperbolic relationship exists between beta-cell function and insulin sensitivity. With deviation from this hyperbola, deterioration of glucose tolerance. Abbreviations: IGT, impaired glucose tolerance; NGT, normal glucose tolerance; T2DM, type 2 diabetes mellitus.
Insulin resistance is strongly associated with obesity for which several mechanisms have been invoked. A number of circulating hormones, cytokines and metabolic fuels, such as nonesterified (free) fatty acids (NEFA) originate in the adipocyte and diminish insulin action (see below). In obese subjects, adipocytes are large, which renders them resistant to the ability of insulin to suppress lipolysis, especially in visceral or deep subcutaneous fat.
This results in elevated release and circulating levels of NEFA and glycerol, both of which aggravate insulin resistance in skeletal muscle and liver (Fig. 2).
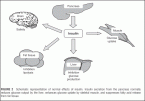 FIGURE 2 Schematic representation of normal effects of insulin. Insulin secretion from the pancreas normally reduces glucose output by the liver, enhances glucose uptake by skeletal muscle, and suppresses fatty acid release from fat tissue.
FIGURE 2 Schematic representation of normal effects of insulin. Insulin secretion from the pancreas normally reduces glucose output by the liver, enhances glucose uptake by skeletal muscle, and suppresses fatty acid release from fat tissue.
Tissue-Specific Insulin Receptor Knockout Models
In order to better understand whether severe insulin resistance can lead to frank diabetes, and to delineate the roles of the various insulin-sensitive tissues, the question has arisen whether it would be possible to induce diabetes in animal models harboring genetic defects leading to absence of insulin receptors in specific tissues (brain, muscle, liver, adipocytes, and pancreas islets).
Therefore, animal models with “conditional knockouts of the insulin receptor” have been studied (Fig. 3). Although it had previously been widely thought that muscle insulin resistance would lead to diabetes, muscle-specific knockout models did not develop diabetes, even if these animals did become obese. Similarly, neither adipocyte-specific or brain-specific insulin receptor knockout animal models became diabetic (Fig. 2), although brain-specific animal models did become overweight, pointing to the physiological role of insulin as a (hypothalamic) satiety factor. However, liver-specific (11) and pancreas beta-cell-specific knockout models did develop diabetes. The latter findings point to the great importance of insulin signaling within the beta cells for beta-cell growth and function.
{kind=link}
However, the main positive conclusion of these studies is that even severe insulin resistance at the level of brain, muscle, or adipocyte does not lead to frank diabetes mellitus.
The finding that severe insulin resistance in the liver or in the pancreas beta cells can lead to frank diabetes in itself cannot be taken as proof of the hypothesis that one or the other is an obligate part of the sequence of events of development of (human) type 2 diabetes.