Insulin Resistance
Cellular Mechanisms
The insulin receptor is specific plasma membrane receptor with tyrosine kinase activity, to which insulin is bound. This kinase activates the insulin receptor substrate (IRS) proteins on multiple sites; these IRS proteins serve as binding scaffolds for a variety of adaptor proteins and lead to the downstream signaling cascade (Fig. 4). Insulin activates a series of lipid and protein kinase enzymes linked to the translocation of glucose transporters to the cell surface, synthesis of glycogen, protein, mRNAs and to nuclear DNA that influences cell survival and proliferation.
Insulin resistance presumably results from mechanisms blocking insulin signaling. It is of note that various normal biological processes can inhibit IRS protein activity via phosphorylation at specific serine and threonine residues within the IRS proteins.
Other processes can interfere with insulin signaling by interfering with other proteins further downstream of the IRS proteins (for example, PKB/akt). Recent research indicates that several of these mechanisms underlie “insulin resistance.”
 FIGURE 3 Schematic representation of effects of animal-tissue specific insulin receptor knockout models.
FIGURE 3 Schematic representation of effects of animal-tissue specific insulin receptor knockout models.
The positive effects on downstream responses exerted by tyrosine phosphorylation of the receptor and the IRS proteins are opposed by dephosphorylation of these tyrosine side- chains by cellular protein-tyrosine phosphatases (PTPs) and by protein phosphorylation on serine and threonine residues (which often occur together). PTP1B is a widely expressed PTP, which has been shown to play an important role in the negative regulation of insulin signaling.
Serine/threonine phosphorylation of IRS-1 reduces its ability to act as a substrate for the tyrosine kinase activity of the insulin receptor and inhibits its coupling to its major downstream effector systems.
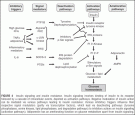 FIGURE 4 Insulin signaling and insulin resistance. Insulin signaling involves binding of insulin to its receptor followed by a cascade of intracellular events, depicted as activation pathways. Negative modulation of insulin action can be mediated via various pathways leading to insulin resistance: Various inhibitory triggers influence their respective signal modulators (partly via transcription factors), which lead via deactivating pathways (tyrosine phosphatases, serine kinases, lipid phosphatases, and degradation pathways) to inhibitory actions on insulin signaling (activation pathways). Adiponectin has an ameliorating function on glucose metabolism apart from insulin signaling.
FIGURE 4 Insulin signaling and insulin resistance. Insulin signaling involves binding of insulin to its receptor followed by a cascade of intracellular events, depicted as activation pathways. Negative modulation of insulin action can be mediated via various pathways leading to insulin resistance: Various inhibitory triggers influence their respective signal modulators (partly via transcription factors), which lead via deactivating pathways (tyrosine phosphatases, serine kinases, lipid phosphatases, and degradation pathways) to inhibitory actions on insulin signaling (activation pathways). Adiponectin has an ameliorating function on glucose metabolism apart from insulin signaling.
Multiple IRS serine kinases have been identified, including various mitogen-activated protein kinases (MAPK/ERK), c-Jun NH3-terminal kinase (JNK), atypical protein kinase C, phosphatidylinositol 3’-kinase, among others. Signal down- regulation can also occur via internalization and loss of the insulin receptor from the cell surface and degradation of IRS proteins.
Members of the “suppressor of cytokine signaling” (SOCS) family of proteins participate in IRS protein degradation through a ubiquitin proteosomal pathway.
Role of Adipocyte Products and Inflammation
Increased levels of NEFA and inflammatory cytokines (e.g., tumor necrosis factor a [TNFa] and interleukin 6 [IL-6]) released by expanded visceral adipose tissue adversely influence the insulin signaling cascade. NEFA inhibit insulin-stimulated glucose metabolism in skeletal muscle and suppress glycogenolysis in liver. NEFA activate cellular kinases, including atypical protein kinase C isoforms by increasing cellular diacylglycerol levels, which can activate the inflammatory kinases inhibitor kB kinase (IKK) and JNK, increasing serine/threonine phosphorylation of IRS-1 and reducing downstream IRS-1 signaling, as described above. TNFa enhances adipocyte lipolysis, which further increased NEFA levels, and also elicits its own direct negative effects on insulin signaling pathways.
Neutralization of TNFa dramatically reverses insulin resistance in rodent models; however, the magnitude of its involvement in human insulin resistance is not entirely clear. The proinflammatory IL-6 inhibits the insulin signal by augmenting the expression of SOCS proteins.
While circulating NEFA and several adipokines are increased in visceral obesity, the levels of the adipose-specific protein adiponectin are decreased, reducing its insulin- sensitizing effects in liver and muscle. Adiponectin signals via AMP kinase, a stress- activated signaling enzyme implicated in a variety of metabolic responses, including suppression of hepatic gluconeogenesis, glucose uptake in exercising skeletal muscle, fatty acid oxidation, and inhibition of lipolysis, which may explain its beneficial metabolic effects.
A close connection between insulin resistance and classical inflammatory signaling pathways has also been recently identified. NF-kB is held in an inactive state in resting conditions by binding to an inhibitory partner, IkB. Phosphorylation of IkB by its kinase (IKK) leads to I B degradation, releasing NF-kB for translocation to the nucleus where it can influence the transcription of diverse genes involved in the inflammatory response. High doses of salicylates, which block IKK activity can ameliorate hyperglycemia and insulin resistance in diabetes and obesity.
More importantly, genetic disruption of IKKb-normalized skeletal insulin resistance caused by NEFA via improvement in IRS-1 tyrosine phosphorylation and activation of its downstream signal cascade. Overall, this line of evidence suggests that IKK may be an important target for the development of new therapeutics in insulin resistance, especially in the setting of visceral adiposity.
In addition to their effects on insulin signaling, the circulating adipose tissue factors strongly influence vascular endothelial function, linking the increased vascular risk in the metabolic syndrome with mechanisms of cellular insulin resistance. Adipose secretory factors also recruit and activate inflammatory cells, which can further perpetuate a systemic inflammatory milieu that can strongly influence vascular function and atherogenesis.
Mitochondrial Metabolism
The accumulation of “ectopic” triglyceride in visceral depots has suggested a defect in mitochondrial lipid oxidation in patients with type 2 diabetes, who have impaired oxidative capacity and small mitochondria in skeletal muscle.
Peroxisome proliferator-activated receptor-gamma (PPAR-γ) coactivator 1 (PGC-1), a transcription factor for genes involved in mitochondrial fatty acid oxidation and ATP synthesis, was decreased in young, lean, insulin- resistant offspring of parents with type 2 diabetes, suggesting that an inherited defect in mitochondrial oxidative phosphorylation could lead to cellular lipid accumulation.
Gene expression profiling studies have also shown that decreased expression of PGC-1 and related gene products may affect mitochondrial function in subjects with insulin resistance and type 2 diabetes.
Michael Stumvoll
Department of Medicine, University of Leipzig, Leipzig, Germany
Barry J. Goldstein
Division of Endocrinology, Diabetes and Metabolic Diseases, Department of Medicine, Jefferson Medical
College of Thomas Jefferson University, Philadelphia, Pennsylvania, U.S.A.
Timon W. van Haeften
Department of Internal Medicine, University Medical Centre Utrecht, Utrecht, The Netherlands
REFERENCES