Positional approaches - The Genetics of Human Obesity
Positional genetic techniques require no special previous knowledge of the function of an individual genomic region but implicate it in the causation of obesity purely on the grounds that identifiable markers in the region are found in obese phenotypes more frequently than would be expected by chance (i.e. they segregate in obesity and are identified in families by linkage or in populations by genetic association).
In practice, there is often a sequence of investigation using positional (described below) and then candidate approaches termed the ‘positional candidate approach’.
After identification of a region of interest using linkage studies in families, the genomic area of interest may be ‘fine mapped’ using techniques such as linkage disequilibrium (LD) to an area a few tens or hundred thousand base pairs in length (Figure 2.2). Subsequent association studies may attempt direct detection of an increased prevalence of a mutation (signalled either by a marker close to the mutation or a specific mutation itself) in affected individuals (probands) relative to unaffected population controls. Candidate studies then focus on genes with a plausible role in body weight regulation in the genomic region thus identified.
This ‘positional candidate’ approach may become more productive in its application to complex polygenic disease following publication of the human genome sequences. Functional annotation of the human genome sequence, now underway, may speed identification of possible candidate genes in stretches of genomic DNA identified by positional approaches. Furthermore, recent enhancements of single nucleotide polymorphism maps (Sachidanandam et al., 2001) will provide many more identifiable genetic markers than have been available hitherto and may thus confer greater precision to gene mapping. 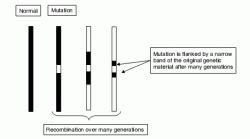
Figure 2.2 Linkage disequilibrium. A disease mutation initially arising on an ancestral haplotype becomes flanked by progressively smaller remnants of the original haplotype that are in LD with it and thus tend not to become separated from it by recombination events. These remnants may contain sequences which can be used to mark the position of the disease locus in a later generation. Although the detection of sequences derived from the ancestral haplotype may become more difficult after many generations, the precision of localization may be increased.
Once identified, further evidence for the potential role of a particular candidate gene in regulation of body weight may be sought from manipulation of the gene in animal models (e.g. transgenic or knockout models). Indeed, where the function of a locus revealed in association or LD studies is poorly understood, this may be vital in order to show that the proposed mutation is causative rather than simply being itself in linkage equilibrium with the real disease locus.
Warden CH and Fisler JS
Katsanis N, Beales PL, Woods MO